AI医療文書レビューの実践ガイド ― 治験・添付文書・安全性情報の品質を効率的に担保する方法
治験文書、添付文書、安全性報告の品質管理にAIを活用する方法を解説。GxP環境でのAIバリデーション要件、リスクベースの導入アプローチ、具体的な活用シーンと注意点を紹介します。
「この治験総括報告書の150ページ、いつまでにレビューを完了できますか?」――医薬品の開発現場で、こうした問いは日常的に発生します。 治験文書の品質レビューは、患者の安全と医薬品の承認に直結する極めて重要な業務です。同時に、膨大なページ数と厳格な品質基準が求められるため、レビューの工数は増大し続けています。
製薬業界では、1つの新薬の申請に必要な文書量がCTD(医薬品申請資料のフォーマット)で数万ページに達することもあります。これらの文書すべてについて、用語の統一性、数値の整合性、規制要件への準拠を人間だけで確認する従来のアプローチは、限界に近づいています。
この記事では、治験文書・添付文書・安全性情報のレビューにAIを活用する方法を、GxP(医薬品・医療機器の品質管理基準)環境でのバリデーション要件も含めて解説します。
医療文書レビューの現状と課題
医療文書のレビュー業務は、他の業界と比べて3つの特殊性があります。
特殊性1: 規制に基づく品質要件
医療文書は、単なるビジネス文書とは異なり、法令や規制ガイドラインに準拠した品質が求められます。GCP(医薬品の臨床試験の実施の基準)、GMP(医薬品及び医薬部外品の製造管理及び品質管理の基準)、GLP(医薬品の安全性に関する非臨床試験の実施の基準)――いわゆるGxPの各基準が、文書の作成・レビュー・保管のすべてに適用されます。
具体的には、「誰が、いつ、何をレビューし、どのような判断をしたか」の記録(監査証跡)が求められます。AIを導入する場合も、この監査証跡の要件を満たす設計が必要です。
特殊性2: 文書間の整合性
医療文書は単体ではなく、複数の文書が相互に参照し合う構造を持っています。治験実施計画書(プロトコル)の記載内容と治験総括報告書(CSR)の結果が一致しているか、添付文書の「効能・効果」と承認条件が整合しているか――こうした文書間の整合性チェックが、レビューの大きな工数を占めています。
ある大手製薬企業の調査では、レビュー工数の約40%が「文書間の整合性確認」に費やされていると報告されています。この領域は、AIの文書横断検索と比較機能が大きな効果を発揮する分野です。
特殊性3: 専門用語と表記の統一性
医療文書では、医学用語の正確さと表記の統一性が厳密に求められます。MedDRA(ICH国際医薬用語集)に基づく有害事象名の表記、化学物質名の記載方法、統計用語の使い方など、専門的な表記ルールが多岐にわたります。
表記ゆれ(例: 「ヘモグロビン」と「Hb」の混在、「副作用」と「有害事象」の使い分け)を人間が数万ページにわたって確認するのは、現実的には困難です。医療・製造の審査プロセスでLLMを安全活用する方法でも触れていますが、こうした網羅的なチェックはAIの得意領域です。
図2: 医療文書レビューの3つの特殊性
AIが活躍する医療文書レビューの5つの領域
医療文書のレビューにAIを適用する際、すべての工程にAIを導入するのではなく、効果が高い領域から段階的に始めるのが現実的です。私たちは、AI適合度が高い順に5つの領域を整理しました。
領域1: 表記統一チェック(AI適合度: 最高)
MedDRA用語、化合物名、統計用語、略語の定義と使い方が文書全体で統一されているかをAIでチェックします。
この領域がAI適合度が最も高い理由は、チェック基準が完全にルール化できるためです。MedDRAの用語辞書、社内の表記ガイドライン、スタイルガイドをAIに参照させれば、数万ページの文書でも一貫した基準でチェックできます。
領域2: 数値・データの整合性チェック(AI適合度: 高)
治験の各相で報告されている数値データ(被験者数、有効率、有害事象の発現率など)が、文書内で一貫しているかをチェックします。
たとえば、「本文で被験者数200名と記載されているのに、表では198名になっている」「方法の章でp値の有意水準を0.05と定義しているのに、結果の章でp=0.048を有意ではないと解釈している」といった不整合を検出します。
領域3: 安全性情報の一次スクリーニング(AI適合度: 高)
ICSR(個別症例安全性報告)の一次スクリーニングは、AI導入の効果が最も実感しやすい領域です。有害事象報告の内容を読み取り、「重篤性の判定」「因果関係の有無の初期判断」「MedDRAコーディング」をAIが一次的に行い、人間のファーマコビジランス(医薬品安全性監視)担当者が確認・承認する流れです。
ICSRの処理件数は新薬発売後に急増するため、AI活用による処理効率の改善効果が大きくなります。AIによる一次スクリーニングで、人間のレビュー時間を40〜60%短縮できたとの報告があります。
領域4: 規制文書のテンプレート準拠チェック(AI適合度: 中)
CTD(コモン・テクニカル・ドキュメント)やeCTD(電子版CTD)のフォーマットに準拠しているかのチェックです。ICH(医薬品規制調和国際会議)のガイドラインに沿ったセクション構成になっているか、PMDA(独立行政法人 医薬品医療機器総合機構)の提出要件を満たしているかをAIが確認します。
この領域は、テンプレートの構造チェック(セクションの有無、順序)はAIが得意ですが、内容の適切性(「この試験デザインはPMDAが求める水準か」)の判断は人間が行う必要があります。
領域5: 文献レビューの効率化(AI適合度: 中)
申請資料に引用する文献のレビューも、AIで効率化できる領域です。関連文献の網羅的検索、文献の要約生成、引用の正確性チェック(文献の内容と引用箇所の整合性)をAIが支援します。
ただし、文献の質の評価(バイアスリスクの判定、エビデンスレベルの判断)は、専門家による判断が不可欠です。
GxP環境でのAIバリデーション: CSAアプローチ
医療文書のレビューにAIを導入する際、最も重要かつ独特な要件がバリデーション(検証)です。GxP環境で使用するコンピュータシステムは、その品質が保証されていることを文書化する必要があります。
従来のCSV(コンピュータ化システムバリデーション)
従来のCSVアプローチでは、GAMP 5(Good Automated Manufacturing Practice)のカテゴリ分類に基づき、すべての機能について詳細なテスト文書(IQ/OQ/PQ)を作成する必要がありました。このアプローチをAIシステムにそのまま適用すると、AIの出力が非決定的(同じ入力に対して毎回同じ出力が保証されない)であるため、従来のバリデーション手法では対応が困難です。
CSA(Computer Software Assurance)への移行
FDAが2022年に公表したCSAドラフトガイダンスは、「リスクに基づいたアプローチ」を推奨しています。2025年以降、この考え方が製薬業界で広く採用されるようになりました。
CSAの核心は「患者安全への影響度に応じて、検証の深さを変える」という考え方です。
高リスク(患者安全に直接影響): AIの判断が承認判断に直結する場合。厳密なバリデーションが必要で、AIの出力を人間が全件レビューする設計にします
中リスク(品質に影響するが安全性への直接影響は限定的): 表記統一チェックや整合性確認など。AIの精度を統計的に検証し、定期的な再検証を行います
低リスク(補助的な利用): 文献検索、要約生成など。基本的な動作確認で十分です
AIのバリデーションはCSAアプローチで
AI/MLシステムに従来のCSVアプローチを機械的に適用するのは非効率です。リスクに応じた検証深度を設定するCSAアプローチを採用し、「何をどこまで検証するか」を事前に文書化してください。PMDAも2024年以降、リスクベースのアプローチを容認する姿勢を示しています。
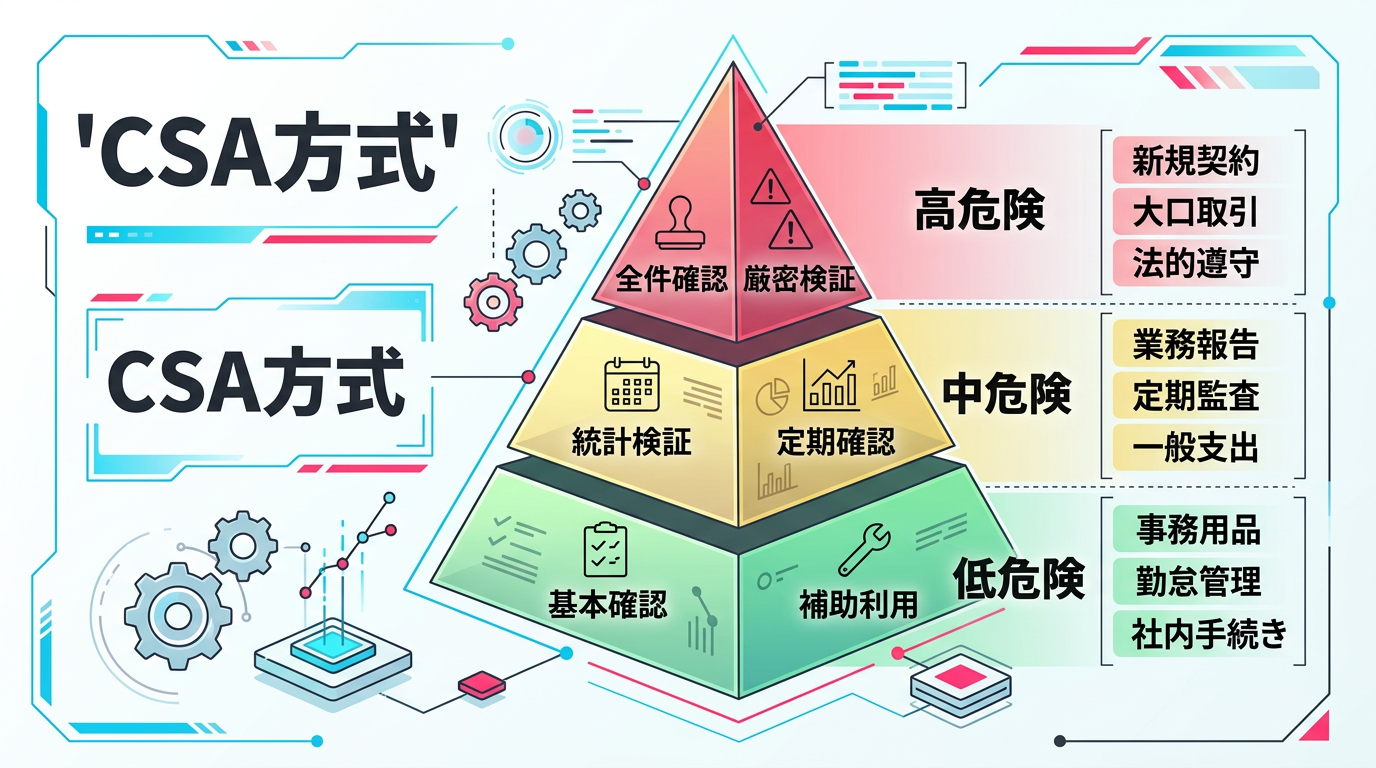
図3: リスクベースのバリデーションピラミッド
導入のための3つの実践ステップ
医療文書レビューへのAI導入は、規制環境の特殊性から慎重に進める必要があります。以下の3ステップで段階的に導入します。
ステップ1: パイロット対象の選定と環境構築
まず、AI適合度が高い「表記統一チェック」または「ICSRの一次スクリーニング」をパイロット対象として選びます。
環境構築では、PHI(保護対象保健情報)の取り扱いが最優先事項です。外部のLLMサービスを使う場合は、データの仮名化(マスキング)処理を前段に組み込みます。AI審査のセキュリティ設計で解説しているアーキテクチャパターンのうち、医療文書では「マネージドVPC環境」または「オンプレミス環境」が推奨されます。
Azure OpenAI Service on your data(社内データを使ったRAG構築)やAmazon Bedrock(VPC内でのモデル利用)は、データが外部に送信されない設計であるため、GxP環境との親和性が高いです。
ステップ2: バリデーション計画の策定と実行
CSAアプローチに基づくバリデーション計画を策定します。計画には以下の項目を含めます。
- 意図する使用目的: AIの出力を何に使うか、最終判断は誰が行うか
- リスクアセスメント: 患者安全への影響度の評価
- 検証方法: 精度の測定方法、サンプルサイズ、合格基準
- 変更管理: AIモデルの更新時の再検証手順
- 定期レビュー: 四半期ごとの精度モニタリング方法
バリデーションの実行では、過去の審査済み文書を「正解データ」として使い、AIの出力と比較します。精度指標として、検出率(見逃さない力)と適合率(誤検出しない力)を算出し、事前に設定した合格基準と照合します。
ステップ3: SOP整備と本番運用
標準作業手順書(SOP)を新規作成または改訂し、AIを活用したレビューフローを正式な業務プロセスとして定義します。SOPには、AIの使用条件、禁止事項、エスカレーションルール、トレーニング要件を明記します。
本番運用では、監査証跡の完全性が特に重要です。「AIがどの文書に対してどのようなチェックを行い、どのような結果を出力したか」「人間がAIの出力をどのように確認・修正したか」のすべてをログとして記録・保管する仕組みを構築します。
文書管理AIで審査プロセスを効率化する方法で解説している監査証跡の設計原則が、医療文書の領域でも適用できます。
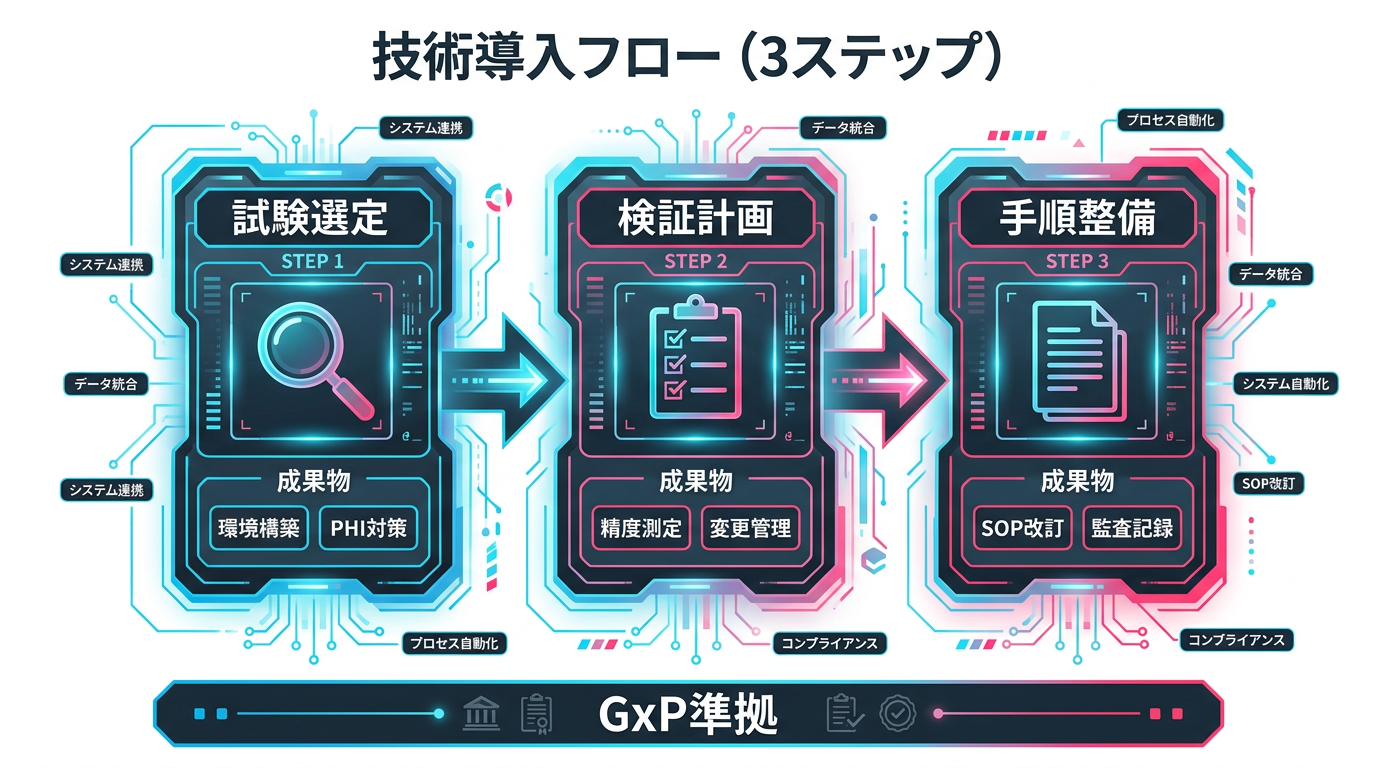
図4: GxP環境でのAI導入3ステップ
よくある質問(FAQ)
GxP環境でAIを使っても規制上問題ないですか?
問題ありません。ただし、AIシステムの使用目的とリスクに応じたバリデーション(検証)が必要です。FDAやPMDAも、リスクベースアプローチによるAI/ML活用を認める方向でガイダンスを整備しています。AIの出力を最終判断とせず、人間による確認を経る設計にすることが基本です。CSA(Computer Software Assurance)のフレームワークに沿って、リスクに応じた検証計画を策定してください。
医療文書のAIレビューで最初に取り組むべき領域は?
安全性情報の個別症例報告(ICSR)の一次スクリーニングが最適です。定型フォーマットで処理件数が多く、チェック基準が明確であるため、AIの精度が出やすい領域です。次に取り組む領域としては、表記統一チェック(MedDRA用語の準拠確認)が推奨されます。いずれも、人間による最終確認を維持しつつ、AIで一次処理の効率化を図るアプローチです。
AIに医療文書を入力する際のセキュリティ上の注意点は?
患者の個人情報(PHI)の取り扱いが最重要課題です。外部のLLMサービスを使う場合は、PHIをマスキング(仮名化)してから入力する必要があります。患者名、ID、生年月日、施設名などの識別情報を自動でマスキングする前処理パイプラインを構築してください。オンプレミス環境やVPC内でのモデル利用も選択肢に含め、自社のセキュリティポリシーに合った方法を選んでください。
医療文書のAIレビューにはどんなツールが使えますか?
製薬業界向けには、Veeva Vault QualityDocやMasterControl等の品質管理システムにAI搭載の文書比較・整合性チェック機能が追加されています。汎用LLMをカスタマイズする場合は、Azure OpenAI Service等のエンタープライズ向けサービスをVPC内で利用するのが一般的です。RAG(検索拡張生成)で社内の規制ガイドラインやSOPを参照させることで、規制要件に特化したチェックが可能になります。
まとめ
医療文書のAIレビューは、規制環境の特殊性を理解した上で、リスクベースのアプローチで段階的に導入することが成功の鍵です。
- 5つの適用領域: 表記統一チェック(最高適合度)→ 数値整合性 → ICSR一次スクリーニング → テンプレート準拠 → 文献レビューの順に導入を検討
- GxPバリデーション: CSAアプローチで「リスクに応じた検証深度」を設定。従来のCSVを機械的に適用しない
- セキュリティ: PHIのマスキングは必須。VPC内でのモデル利用が推奨
- 監査証跡: AIの入力・出力・人間の確認結果のすべてを記録する仕組みが不可欠
次のアクションとして、自社の医療文書レビュー業務で「表記統一チェック」にどの程度の工数がかかっているかを計測してみてください。月間10時間以上をこの作業に費やしているなら、AIによる自動化の投資対効果は十分に見合います。

この記事の著者
Naosy 編集部
レビュー・校正・審査プロセスの最適化に関する実践的なナレッジを発信しています。



